Demo
Skip right to the code and documentation:
Code: https://colab.research.google.com/drive/1z0ilrakoRJ8maapMNHwtPf83pKK1THST?usp=sharing
Technical Documentation: https://sulstice.gitbook.io/globalchem-your-chemical-graph-network/cheminformatics/decoding-fingeringprints-and-smiles-to-iupac
Philosophy
I classify my data according to a functional group or name that makes sense to me. Most of the time my philosophy is if I can’t speak it then it’s not natural to me. That being said, when I explore the chemical universe I often need a way to remember the functional groups I am going after and if I want to navigate that chemical space then I need to be armed with reference patterns. Should we look at food? narcotics? war? poison? or medicine? you decide. It’s not my choice, I can only send a message. The idea is simple convert this:
00000000000000000000000000000000000000000000000000000000000000001000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000001000000000000000000000000000000000000000000000000000100000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000to this
['benzene', 'ammonia']Hyperparameters Encoding SMILES
I’ve talked about Morgan before and it’s been discussed a lot as an implementation in RDKit. We need to pick standards for fingerprinting and the best one I have found in my experience as a cheminformatician is 512 bit length with a radius of 2. I have found this big enough to capture the chemical environment. If we create a reference standard for fingerprints with common chemical lists we can decode the information readily and allow us to amply explore chemical space with purpose.
bit_string = AllChem.GetMorganFingerprintAsBitVect(
molecule,
2,
nBits=512
).ToBitString()This is the meat of the code and you can reference the RDKit documentation for more. So that’s what I did. Created nearly 3000 bit vectors organized into GlobalChem:

From here to classify a fingerprint was pretty simple to implement with a common similarity score function called Tanimoto Similarity. Where I use a similarity checker and anything bit vector that is above a score of 90% similarity is marked as being a functional group that exists within that space.
You can select a node and then pull a similarity by rebuilding the structure from the reference standard.
fingerprint = DataStructs.cDataStructs.CreateFromBitString(fingerprint)
for smiles, reference_fingerprint in bit_strings.items():
reference_fingerprint = DataStructs.cDataStructs.CreateFromBitString(reference_fingerprint)
score = DataStructs.FingerprintSimilarity(fingerprint, reference_fingerprint)So let’s look at the example in the demo:
Install the Package:
!pip install -q global-chem[cheminformatics] --upgradeLoad the cheminformatics and decoder engine package:
from global_chem import GlobalChem
from global_chem_extensions import GlobalChemExtensionsgc = GlobalChem()
cheminformatics = GlobalChemExtensions().cheminformatics()
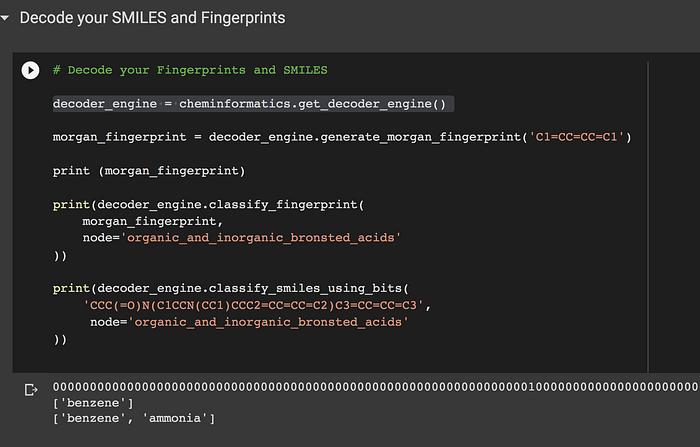
decoder_engine = cheminformatics.get_decoder_engine()
Load the benzene molecule written in SMILES into a Morgan fingerprint:
morgan_fingerprint = decoder_engine.generate_morgan_fingerprint('C1=CC=CC=C1')print(decoder_engine.classify_fingerprint(
morgan_fingerprint,
node='organic_and_inorganic_bronsted_acids'
))
And then using a node that’s not a bad reference standard for functional groups we can retrieve the IUPAC name.
['benzene']So this helps us decode fingerprints by having an annotated reference and safe passage of chemical information as our predecessors before us.
Decoding Bigger SMILES
Now let’s take a more complex example where a SMILES string might be more complex and not have a reference standard directly into GlobalChem. Well we can used the BRICS module implemented in RDKit to fragment molecules and perhaps smaller molecules will have a reference in the fingerprint. Then we can join all the functional groups together to get an idea of the chemical space inside a fingerprint written in english. Let’s take a look at an example of fentanyl:
print(decoder_engine.classify_smiles_using_bits(
'CCC(=O)N(C1CCN(CC1)CCC2=CC=CC=C2)C3=CC=CC=C3',
node='organic_and_inorganic_bronsted_acids'
))Here we can see something more complex and maybe using a small set of functional groups in the bronsted acids is not enough to match compounds in there. So we fragment the molecule:
fragments = list(BRICS.BRICSDecompose(Chem.MolFromSmiles(smiles)))And then convert to fingerprints, loop through for similarity, and there it is:
Hopefully this makes it easy for everyone to understand their fingerprints and for my ai models to learn efficiently.
