A nice and bright Sunday morning to design Convolutional Neural Networks. A CNN is a neural network trained on using images and passed through a series of filters to determine features. As an organic chemist, we use our eyes in designing structures so perhaps this an intuitive machine learning approach and fragment a molecule using our own internal filters. It seems natural to try it out.

Our goal for this network is we want to know if a molecule will behave like cannabis based on it’s binding constant (how strong a ligand binds) into a common endocannabinoid receptor, CB1, a principle target for THC. The crystal structure was derived by Hua et al.
The structure is a series of alpha helices in a shoe type shape with the binding pocket being very deep within the receptor.

The of THC to a CB1 receptor can be quantified with an inhibition constant, Ki, which is an indication into how potent an inhibitor is. Small Ki values of 100 micro molar are considered to be potent.
If we take a list of inhibitors for CB1 and their inhibition constants as recorded in nM from this paper:
Can we predict whether a new cannabinoid compound would behave similar to a terpene like THC based on it’s inhibition constant value. Here’s some of the data abstracted from the paper. Feel free to grab more.
compounds = {
'CCCCC1=CC(=C2C3C=C(CCC3C(OC2=C1)(C)C)C)O': 36,
'CCCC1=CC(=C2C3C=C(CCC3C(OC2=C1)(C)C)C)O': 22,
'CCCCCC1=CC2=C(C3C=C(CCC3C(O2)(C)C)C)C(=C1C(=O)O)O': 620,
'CCCCCC1=CC(=C(C(=C1)O)C/C=C(\C)/CCC=C(C)C)O': 1045,
'CCCC1=CC(=C(C(=C1)O)C/C=C(\C)/CCC=C(C)C)O': 2865,
'CCCCCC1=CC(=C(C(=C1C(=O)O)O)C/C=C(\C)/CCC=C(C)C)O': 13116,
'COC1=C(C/C=C(CC/C=C(C)\C)\C)C(O)=CC(CCCCC)=C1': 10000,
'CCCCCC1=CC(=C(C(=C1)O)C2C=C(CCC2C(=C)C)C)O': 1690,
'CCCC1=CC(=C(C(=C1)O)C2C=C(CCC2C(=C)C)C)O': 14445,
'CCCCCC1=CC(=C(C(=C1C(=O)O)O)C2C=C(CCC2C(=C)C)C)O': 626,
}To design our CNN, we will first need to prepare the data into an image based format. To do so, we will use the theory of “Chemception” where a SMILES string is converted into a 2D molecular coordinates and mapped onto a 80 by 80 grid. Where the atoms and bonds are color coded and everything else is black.

Let’s get our imports going:
import rdkit
import numpy as np
import pandas as pd
from rdkit import Chem
from rdkit.Chem import AllChem
from matplotlib import pyplot as pltAnd convert our data into a Dataframe:
df = pd.DataFrame()
df['smiles'] = list(compounds.keys())
df['ki'] = list(compounds.values())
df["mol"] = df["smiles"].apply(Chem.MolFromSmiles)Next we want to create a chemcepterize function. This first is to create the image space of the molecule. You want a molecule that is centered and not too small and is readable to the network. We call these parameters embed and res . By changing the embedding you can alter the size of the image. Obviously, the embedding where it’s higher might have a poor time or a lot of dropout of nodes since a lot of space is empty.
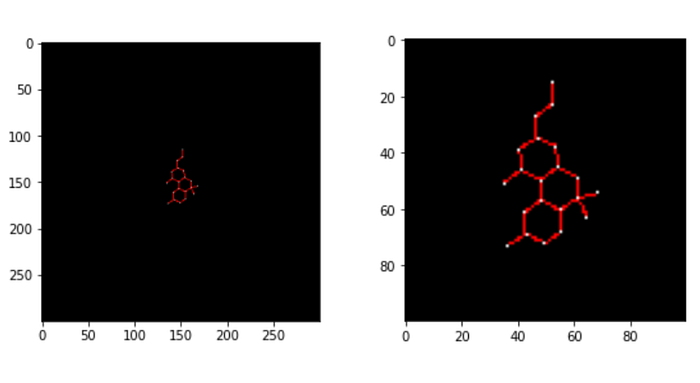
The effect of a resolution on the image can be where the space occupied by different information can dominate.
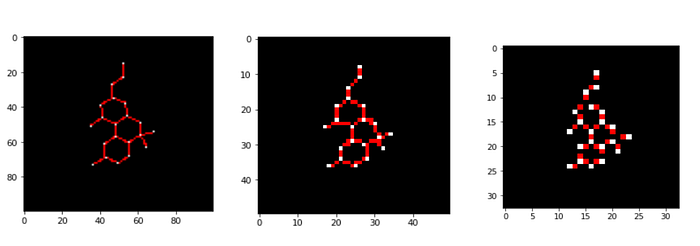
The embedding and resolution of your image is very important because it allows the neural network to abstract features more efficiently. So we compute the dimensions and the 2D coordinates and set up 4 empty zeroes that will act as a meta store for information like atomic number.
def chemcepterize_mol(mol, embed=10.0, res=0.2):
dimensions = int(embed*2/res)
chempcepterized_molecule = Chem.Mol(mol.ToBinary())
chempcepterized_molecule.ComputeGasteigerCharges()
AllChem.Compute2DCoords(chempcepterized_molecule)
coords = chempcepterized_molecule.GetConformer(0).GetPositions()
vector = np.zeros((dims,dims,4))The initial vector looks something like this:
[[[0. 0. 0. 0.]
[0. 0. 0. 0.]
[0. 0. 0. 0.]
...
[0. 0. 0. 0.]
[0. 0. 0. 0.]
[0. 0. 0. 0.]]
[[0. 0. 0. 0.]
[0. 0. 0. 0.]
[0. 0. 0. 0.]
...
[0. 0. 0. 0.]
[0. 0. 0. 0.]
[0. 0. 0. 0.]]
[[0. 0. 0. 0.]
[0. 0. 0. 0.]
[0. 0. 0. 0.]
...
[0. 0. 0. 0.]
[0. 0. 0. 0.]
[0. 0. 0. 0.]]Next, we want to iterate through the molecule’s bonds and embed the bond order information into each pixel at that particular coordinate. To do that, we iterate through each bond and grab the start, end atom index.
for i,bond in enumerate(mol.GetBonds()):
bond_order = bond.GetBondTypeAsDouble()
start_atom_index = bond.GetBeginAtomIdx()
end_atom_index = bond.GetEndAtomIdx()
start_coordinates = coords[start_atom_index]
end_coordinates = coords[end_atom_index]To put the bond order information into the image, we want to translate the numbers into a more manageable dimension that scales with our resolution and built a line between the two. We set up a fractional space between 0 and 1 with the resolution being how many pixels will take up that information. The line will be points a long the space and then supplied the bond order information.
fractional_space = np.linspace(0,1,int(1/res*2))
for fraction in fractional_space:
coordinates = (fraction*start_coordinates + (1-fraction)*end_coordinates)
x = int(round((coordinates[0] + embed)/res))
y = int(round((coordinates[1]+ embed)/res))
vector[ x, y, 0] = bond_orderThe first channel is used to take those coordinates and correlate a bond order number. Let’s do the same thing with the atom and supply the other channels with the atomic_number , hybridization , and gasteiger charge . And finally return the vector.
for i,atom in enumerate(cmol.GetAtoms()):
x = int(round((coords[i][0] + embed)/res))
y = int(round((coords[i][1]+ embed)/res))
vector[ x , y, 1] = atom.GetAtomicNum()
hyptype = atom.GetHybridization().real
vector[ x , y, 2] = hyptype
charge = atom.GetProp("_GasteigerCharge")
vector[ x , y, 3] = charge
return vectOur full function then looks like this:
def chemcepterize_mol(mol, embed=10.0, res=0.2):
dimensions = int(embed*2/res)
chempcepterized_molecule = Chem.Mol(mol.ToBinary())
chempcepterized_molecule.ComputeGasteigerCharges()
AllChem.Compute2DCoords(chempcepterized_molecule)
coords = chempcepterized_molecule.GetConformer(0).GetPositions()
vector = np.zeros((dims,dims,4))
# Bonds
for i,bond in enumerate(mol.GetBonds()):
bond_order = bond.GetBondTypeAsDouble()
start_atom_index = bond.GetBeginAtomIdx()
end_atom_index = bond.GetEndAtomIdx()
start_coordinates = coords[start_atom_index]
end_coordinates = coords[end_atom_index]
# Draw Bond A long Vector
fractional_space = np.linspace(0,1,int(1/res*2))
for fraction in fractional_space:
coordinates = (fraction*start_coordinates + (1-fraction)*end_coordinates)
x = int(round((coordinates[0] + embed)/res))
y = int(round((coordinates[1]+ embed)/res))
vector[ x, y, 0] = bond_order
# Atoms
for i,atom in enumerate(cmol.GetAtoms()):
x = int(round((coords[i][0] + embed)/res))
y = int(round((coords[i][1]+ embed)/res))
vector[ x , y, 1] = atom.GetAtomicNum()
hyptype = atom.GetHybridization().real
vector[ x , y, 2] = hyptype
charge = atom.GetProp("_GasteigerCharge")
vector[ x , y, 3] = charge
return vectOur final function then could look like this for our DataFrame:
def vectorize(mol):
return chemcepterize_mol(mol, embed=12)
df["mol_image"] = df["mol"].apply(vectorize)Now that our data is prepared, we will build the CNN. Try preparing the data yourself and getting a feel for chemcepterizing a list of molecules.
