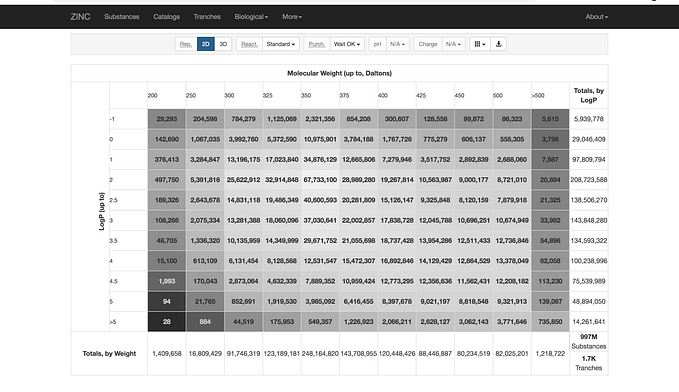
Organic Chemistry
This came at the request of a redditor to teach stereochemistry in SMILES and I completely forgot about how important this was to teach at the beginning. It’s good to ask questions, helps me know what to teach. So let’s go back to a simple stereoisomer, valine:

Where the amine is sticking out from the page which if you remember from organic chemistry we mark with a thick wedge. The priority of the functional groups then comes into play with the tertiary carbon that the purple arrow is highlighting.
Let’s look into the bond connections from that carbon:
1. Carbon - Nitrogen
2. Carbon - Carbon
3. Carbon - Carbon
4. HydrogenSo since Nitrogen is higher priority we mark it with a 1. Then we look at the next set of connections and then reduce:
2. Carbon - Carbon - Oxygen
3. Carbon - Carbon - Carbon
4. HydrogenSo now the carboxylic group is priority number 2. The isopropyl group is 3, and the hydrogen is placed 4th (not shown) going towards the back. In organic chemistry, we can denote the 3D of a molecule if the substituent is going back by either Rectus meaning clockwise, R , or Sinister meaning anti-clockwise, S. If we follow the purple arrow in Figure 1, we are going in reverse so we denote this configuration of Valine as S. If the hydrogen was pointing forward we would be doing R.
In SMILES, we it’s important to preserve this type of information when recording to the computer. We record the SMILES of R with @@ and S with @ . We mark the hydrogen to tell if it is point forward or back attached to the heteroatom with a [] For example:
[C@H] # Hydrogen Pointing Back
[C@@H] # Hydrogen Pointing ForwardSo we if write our valine in it’s isomeric form we achieve:
valine = 'CC([C@H](N)C(O)=O)C'Programming
Let’s crank out the Rdkit module and see if we can see something in the code in generating a stereoisomer. First our imports, and we will be using the EnumerateStereoisomers module. First we pass in our non-isomeric form
from rdkit import Chem
from rdkit.Chem import EnumerateStereoisomers
root_smiles = 'CC(C(N)C(O)=O)C'
molecule = Chem.MolFromSmiles(root_smiles)Next we set it up where we generate all possible stereoisomers for valine where we only want uniqueSMILES. The tryEmbedding flag tests whether the 3D stereoisomer is geometrically possible.
options = EnumerateStereoisomers.StereoEnumerationOptions(unique=True, tryEmbedding=True)
isomers = tuple(EnumerateStereoisomers.EnumerateStereoisomers(
molecule,
options=options)
)And finally we generate the SMILES for all the molecules:
for smiles in isomers:
print(Chem.MolToSmiles(smiles, isomericSmiles=True))Which then returns our SMILES strings:
CC(C)[C@H](N)C(=O)O
CC(C)[C@@H](N)C(=O)OHere we can see all possible combinations are generated and maybe to get to our valine we can filter out the list for only fetching strings with 1 @ or there could be a variety of things there. I hope this was meaningful at understanding stereochemistry in SMILES and where it plays a role.
Happy Cheminformatics!