It’s time to revisit some Molecular Dynamics and refresh myself in some of my own tutorials.
We are going to have a look at a Tyrosine Kinase and it’s inhibitors and simulate the protein movement with CHARMM Force Fields using the Amber Molecular Dynamics Engine. Our goal is to get an understand of how the protein moves in solution and get an idea of it’s behaviour and the binding pocket.
You will need the academic free version of CHARMM and to register your account. We will be writing CHARMM input files for half of this before we switch on over to Amber.
To visualize a PDB file, I use Chimera from UCSF and it runs happily on a apple laptop. The PDB file is provided in the repository:

This abstracted from the crystal structure. In Molecular Dynamics, we move through a series of protocols to ensure that we simulate proteins correctly and consistently. This prevents from odd artifacts showing up or your protein unraveling or even worse….exploding.
Step 1: Add Dummy Hydrogens to the Protein, Minimize and Write the PSF and CHARMM CRD file.
Let’s first read in the PDB file as provided in the software repository. Let’s have a look at the run.inp file in which we will be doing our reading of the protein, adding hydrogens, minimizing them, and writing the output files.
* ADD dummy H for protein
random uniform
faster onWe want to setup two things in CHARMM first. random uniform meaning we can set how we want to distribute the numbers when making estimations or calculations. This could potentially change the numbering search space.
faster on means that in CHARMM there is slow and fast methods for estimating the free energy of bonds, angles, dihedrals, impropers and nonbonded terms when doing the optimization process. There is a trade off in accuracy. Because we want to not kill our hardware and perform numerical operations on what we have. We choose to turn it on. More can be found over at their documentation.
Next, we read in the files and set thesegment ID of the protein, toppar files, and coordinate information.
! Inputs and Variables
!-------------------------
set in = pdb_files/model
set out = results/step1_out_model
proteinset segi = PROA ! segname ID of protein
! Read topology and parameter files
!----------------------------------
stream toppar.str
! Read Sequence Information
!---------------------------
open read card unit 1 name @{in}.pdb
read sequence pdb unit 1 resid
generate PROA setup
close unit 1
! Read Coordinate Information
!----------------------------
open read unit 1 form name @{in}.pdb
read coor pdb unit 1 resid
close unit 1Next we want to generate the internal coordinates using the parameter values from CHARMM.
ic para
ic buildBuild the hydrogens and generate the angles/dihedrals:
!!H-Build
!--------
HBuild
AUTOGEN ANGLes DIHEdAfter the hydrogens are added to the protein we want to them perform a minimization where we want the hydrogens to be in their most favorable state by calculating the derivative of the potential energy and using that information to estimate a lower energy.
Steepest Descent (SD) is a method in which it can rapidly fix molecules that are in a bad conformation. However, it lacks in reaching a convergence and achieving an optimal state. We apply it first to fix and bad changes.
We use the Newton-Raphson method (ABNR) which can recognize and avoid saddle points in a minimization allowing it to achieve a minima.
! Minimize hydrogens ONLY
! -----------------------
cons fix sele .not. hydrogen end ! fix heavy atoms
mini sd nstep 50 nprint 5
mini abnr nstep 10 nprint 1
cons fix sele none end ! MUST remove fix
cons harm force 5.0 sele .not. hydrogen end ! restrain heavy atoms
mini sd nstep 50 nprint 5
mini abnr nstep 10 nprint 1
cons harm clear ! remove restraintsWe perform this first on only hydrogens and then for the both with a constraint on the hetereo-atoms to prevent any potential artifacts. The last thing we want to do is write our outputs and generate our PSF and CRD file which contain the charge information and coordinates to be used later on.
open write unit 90 card name @{out}.psf
write psf card unit 90
close unit 90
open write unit 90 card name @{out}.crd
write coor card unit 90
close unit 90
open write unit 90 card name @{out}.pdb
write coor pdb unit 90
close unit 90
stopNext run the CHARMM script:
charmm -i run.inpLet’s look at the protein before and after the minimization:
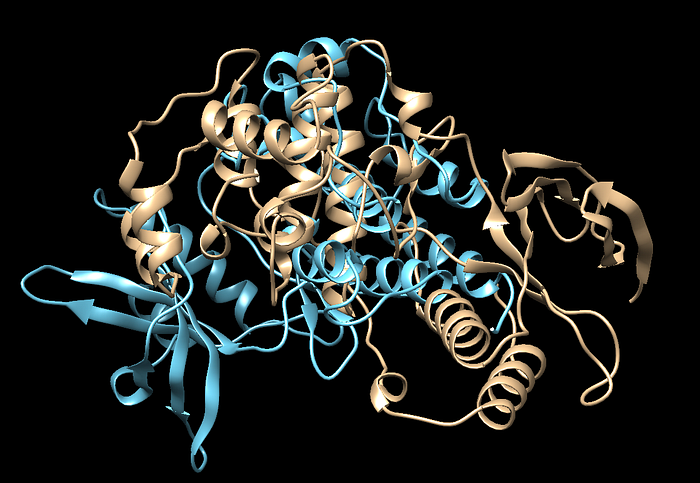
Your protein might look like this so you would want to perform an alignment so we can get a better idea otherwise this is trick to visualize. We have a matchmaker option in Chimera we can use
And then lets have a look at the alignment:
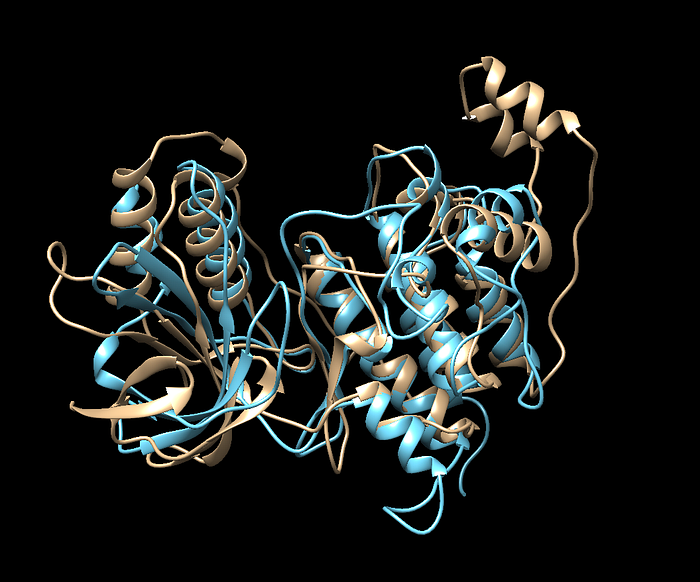
The blue is the protein after minimization and looks like it was more coiled with the alpha helices on the top right. That’s a pretty big significant difference between the two.
Step 1 is Complete if you made it this far! Stay tuned for Step 2.
