Molecular Dynamics (MD) is the study of predicting atomistic movement in the computer as it goes through time and determining whether you can simulate natural forces of the world. For example, in pharmaceuticals and the cheminformatic industry people use MD to simulate the binding of a ligand with a protein.
Thank you to Anu, a student, for completing the installation and setting up the MD.

One of the drawbacks of MD is the computational cost and that itself can start to serve as a bottleneck for common people wanting to do computational chemistry at home.
To ease this we decided to make things easier for the community by teaching them how to install GROMACS on a cloud environment with Github Actions.

Github Actions enables us to run a cloud computer at will. As an organization you are allocated 50,000 minutes per month.

This allows us to have some fun in running simulations at very little cost to the user. To do this we standardized the github actions workflow to install GROMACS that way anyone who is struggling can catch up with ease. First head to the repository:

And then head to the .github workflows tab. In here, you will find the github actions workflow that will install GROMACS and run a MD simulation:

Here you can either fork the repo or copy/paste or request access if you want to come under my umbrella to perform a GROMACS MD simulation.

Click Run GROMACS Simulations:
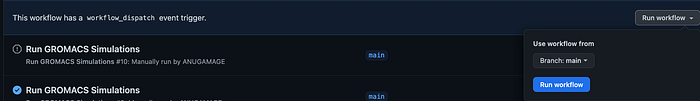
What will happen is a server will spawn on the ubuntu server and a popup will show of the new server.
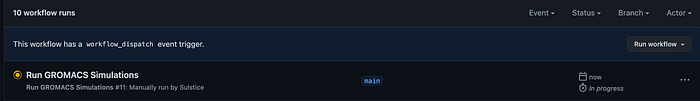
Next go ahead and click the run:
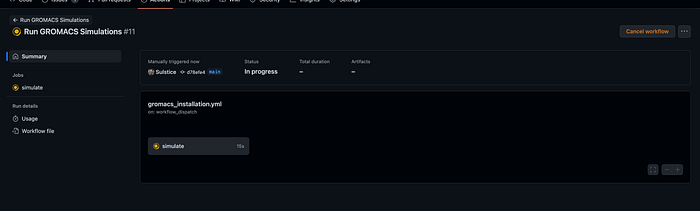
This is the overview of the job, what you can do is click the job to watch the instructions of install GROMACS and then performing a MD simulation.
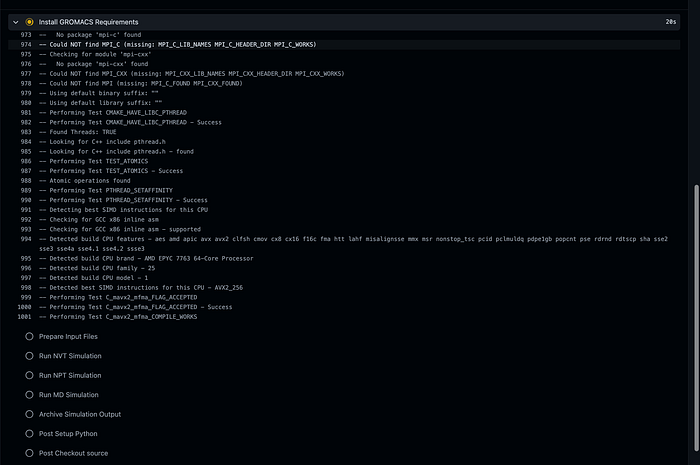
Now, if you don’t have the resources to do this you can sign up for a github account and perform you own github actions code:
For the full action code:
name: Run GROMACS Simulations
on:
workflow_dispatch
jobs:
simulate:
runs-on: ubuntu-latest
timeout-minutes: 240
steps:
- name: Checkout source
uses: actions/checkout@v2
- name: Setup Python
uses: actions/setup-python@v2
with:
python-version: 3.7
- name: Install GROMACS Requirements
run: |
sudo apt-get update
sudo apt-get install -y wget
wget https://ftp.gromacs.org/gromacs/gromacs-2023.3.tar.gz
tar xfz gromacs-2023.3.tar.gz
cd gromacs-2023.3
mkdir build
cd build
cmake .. -DGMX_BUILD_OWN_FFTW=ON -DREGRESSIONTEST_DOWNLOAD=ON
make
make check
sudo make install
echo 'source /usr/local/gromacs/bin/GMXRC' >> $HOME/.bashrc
source $HOME/.bashrc
- name: Prepare Input Files
run: |
cp simulation/input_files/NVT.mdp .
cp simulation/input_files/NPT.mdp .
cp simulation/input_files/MD.mdp .
cp simulation/input_files/EM.gro .
cp simulation/input_files/topol.top .
cp simulation/input_files/index.ndx .
- name: Run NVT Simulation
run: |
gmx grompp -f NVT.mdp -c EM.gro -r EM.gro -p topol.top -n index.ndx -maxwarn 2 -o NVT.tpr
gmx mdrun -deffnm NVT
- name: Run NPT Simulation
run: |
gmx grompp -f NPT.mdp -c NVT.gro -r NVT.gro -p topol.top -n index.ndx -maxwarn 2 -o NPT.tpr
gmx mdrun -deffnm NPT
- name: Run MD Simulation
run: |
gmx grompp -f MD.mdp -c NPT.gro -t NPT.cpt -p topol.top -n index.ndx -maxwarn 2 -o MD.tpr
gmx mdrun -deffnm MD
- name: Archive Simulation Output
uses: actions/upload-artifact@v2
with:
name: simulation-results
path: ./*.{edr,log,trr,tpr,xtc,xvg}We can go into more detail into this code later as we delve into MD but it would be good to get your hands dirty first with running the code.
