Trends in fingerprint matching of peptides using Tanimoto, RDKit, and Cocktail Shaker.
The Peptide Pattern
Peptide therapeutics is something I always have been fond of and lately, I’ve been getting really into converting more peptide tools into the open-source world.
My first exposure was when I decided “Cocktail Shaker” should be primarily a peptide tool instead of something more generic. One of my requirements was that I needed to build a function that generates a SMILES peptide empty “slot” string.
Check out what I mean in this 2D representation:

Each “A” represents an empty slot on the peptide chain where you could perhaps install a natural or non-natural amino acid. This will be the mechanism used to Cocktail Shaker later on but not needed for this blog! Okay, so lets look a little deeper into what a peptide SMILES exactly is, take a tripeptide sequence without the “slots”:
NCC(NCC(NCC(NCC(O)=O)=O)=O)=OAnd it’s 2D

Hmmm what about a 4 length peptide sequence:
NCC(NCC(NCC(NCC(NCC(O)=O)=O)=O)=O)=Oand it’s 2D

Okay so if we keep growing in length we start to see a pattern for a unique representation of a peptide in the SMILES language
3 : NCC(NCC(NCC(NCC(O)=O)=O)=O)=O
4 : NCC(NCC(NCC(NCC(NCC(O)=O)=O)=O)=O)=O
5 : NCC(NCC(NCC(NCC(NCC(NCC(O)=O)=O)=O)=O)=O)=O
6 : NCC(NCC(NCC(NCC(NCC(NCC(NCC(O)=O)=O)=O)=O)=O)=O)=O
7 : NCC(NCC(NCC(NCC(NCC(NCC(NCC(NCC(O)=O)=O)=O)=O)=O)=O)=O)=O
8 : NCC(NCC(NCC(NCC(NCC(NCC(NCC(NCC(NCC(O)=O)=O)=O)=O)=O)=O)=O)=O)=OStart to notice a pattern yet? So originally I took this pattern to build the function PeptideBuilder where I started from the center and built the c_terminus and n_terminus respectfully following the repeated peptide SMILES of the peptide.
n_terminus = "NCC("
c_terminus = "O)=O)"
for i in range(desired_length):
n_terminus += 'NCC('
c_terminus += '=O)'peptide_backbone = n_terminus + c_terminus[:-1]
and voila! a function to build peptide strings.
Peptide Pattern Matching
Here is where it gets a little more interesting, lets cycle back to our original peptide molecule with the slots installed.
Lets use Cocktail Shaker to generate some random molecules of a peptide length of 3 and install Bromine, Fluorine, and Iodine in random slots along the chain. We can use Cocktail Shaker to generate all the combinations.
from peptide_builder import PeptideBuilder
from functional_group_enumerator import Cocktailpeptide_backbone = PeptideBuilder(3)
cocktail = Cocktail(peptide_backbone,ligand_library = ['Br', 'F', 'I'])
combinations = cocktail.shake()
print (combinations)>>>
['NC(F)C(=O)NC(Br)C(=O)NC(I)C(=O)NCC(=O)O', 'NC(Br)C(=O)NC(I)C(=O)NC(F)C(=O)NCC(=O)O', 'NC(I)C(=O)NC(Br)C(=O)NC(F)C(=O)NCC(=O)O', 'NC(Br)C(=O)NC(F)C(=O)NC(I)C(=O)NCC(=O)O', 'NC(I)C(=O)NC(F)C(=O)NC(Br)C(=O)NCC(=O)O', 'NC(F)C(=O)NC(I)C(=O)NC(Br)C(=O)NCC(=O)O']
What does that look like in 2D? Here is an example of the first one. See how it installed Fluorine, Bromine, and Iodine respectfully in the open slots.
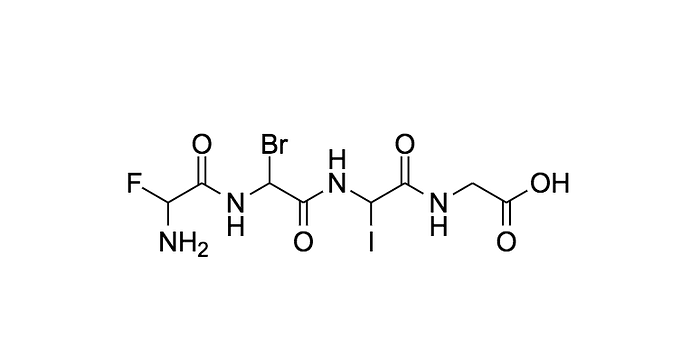
Moving on lets start comparing how similar are the combinations generated to each other?
For example this:
NC(F)C(=O)NC(Br)C(=O)NC(I)C(=O)NCC(=O)O with:
NC(Br)C(=O)NC(I)C(=O)NC(F)C(=O)NCC(=O)OLets see that in 2D: this
with
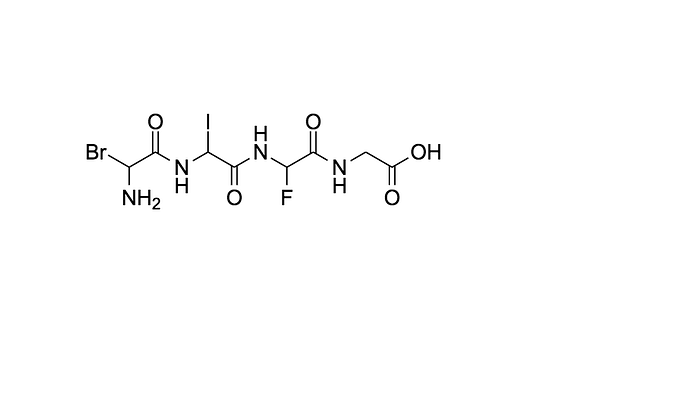
So how do we accomplish this? Luckily there are already some calculated mathematical similarity functions that have been automatically determined for us specifically for cheminformatics! MDL Density, Fragment-based daylight, BCI, Unity 2D (TRIPOS)…all the fun ones, but one particular similarity stood amongst the rest Tanimoto index as a crowd favourite — here’s a quote:
the well-established Tanimoto is the coefficient of choice for computing molecular similarities unless there is specific information about the sizes of the moleculesSo let’s take that as a grain of salt and get some more code flowing to see how we compare these compounds. Let’s make a quick function first the arguments and the doc strings:
First we need the SMILES representation of each molecule (which we have from Cocktail Shaker) and fingerprints which are binary bit strings that represent a pattern within the string.
from rdkit.Chem import AllChem as Chem
from rdkit.Chem.Fingerprints import FingerprintMolspeptide_backbone = PeptideBuilder(3)
cocktail = Cocktail(peptide_backbone,ligand_library = ['Br', 'F', 'I'])
combinations = cocktail.shake()
# Render to molecules
molecules = [Chem.MolFromSmiles(x) for x in combinations]
# Render to fingerprints
fingerprints = [FingerprintMols.FingerprintMol(x) for x in molecules]
Okay, now on to the next step we want to set up a Tanimoto function that will compare one string of the SMILES to the rest of the list and calculate the similarity index
def compare_tanimoto_fingerprints_pairwise(smiles, fingerprints):
query, target, similarity = [], [], []query, target, and similarity will be list stores of comparing one string to another and determining the index.
Next we enumerate through the fingerprint and generate the all different similarities. This might generate combinations than intended but still something interesting to look at
for index, fingerprint in enumerate(fingerprints):
similarity_group = DataStructs.BulkTanimotoSimilarity(
fingerprint, fingerprints[index+1:])
for i in range(len(similarity_group)):
query.append(combinations[index])
target.append(combinations[index+1:][i])
similarity.append(similarity_group[i])Lets wrap it in a dataframe for easier sake to handle the data:
# build the dataframe and sort it
similarity_data = {'query':query,
'target':target,
'similarity':similarity}
similarity_dataframe = pd.DataFrame(data=similarity_data) \
.sort_values('similarity', ascending=False)and presto we get this interesting result:
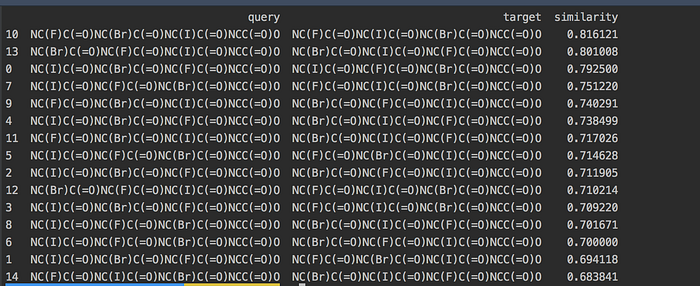
It looks like using Tanimoto Similarity likes a preference to particular atoms which gives it a slightly more similarity. If we compare the top row
# First Row
target : NC(F)C(=O)NC(Br)C(=O)NC(I)C(=O)NCC(=O)O
query : NC(F)C(=O)NC(I)C(=O)NC(Br)C(=O)NCC(=O)O
similarity: 0.816So the fluorine is perhaps weighted more? Let’s compare the second row with bromine:
# Second Row
target : NC(Br)C(=O)NC(F)C(=O)NC(I)C(=O)NCC(=O)O
query : NC(Br)C(=O)NC(I)C(=O)NC(F)C(=O)NCC(=O)O
similarity: 0.801So far F > Br > ? (you think it would be iodine?)
# Third Row
target : NC(I)C(=O)NC(Br)C(=O)NC(F)C(=O)NCC(=O)O
query : NC(I)C(=O)NC(F)C(=O)NC(Br)C(=O)NCC(=O)O
similarity: 0.792Interestingly some halogens are weighted more than others. Where fluorine will have a greater preference and perhaps increasing your score. What other trends can you find?
Update: I read a little more about Tanimoto and found this resource: pdf incredibly helpful.
After fully understanding how Tanimoto works it was pretty clear (with the help of some Linkedin comments) there is a direct influence of how big the atom impacts the score.
Full code you can find here: github or just directly copy and paste from here:
from peptide_builder import PeptideBuilder
from functional_group_enumerator import Cocktail
from rdkit.Chem import AllChem as Chem
from rdkit.Chem.Fingerprints import FingerprintMols
from rdkit import DataStructs
import pandas as pd
import matplotlib
matplotlib.use('PS')
def compare_tanimoto_fingerprints_pairwise(smiles, fingerprints):
"""
Arguments:
smiles (List): List of smiles you would like to compare
fingerprints (List): List of fingerprint RDKit objects for each smiles (should directly correlate)
Returns:
similarity_dataframe (Pandas Dataframe Object): a dataframe containing pairwise similarity.
"""
query, target, similarity = [], [], []
for index, fingerprint in enumerate(fingerprints):
similarity_group = DataStructs.BulkTanimotoSimilarity(fingerprint, fingerprints[index+1:])
for i in range(len(similarity_group)):
query.append(combinations[index])
target.append(combinations[index+1:][i])
similarity.append(similarity_group[i])
# build the dataframe and sort it
similarity_data = {'query':query, 'target':target, 'similarity':similarity}
similarity_dataframe = pd.DataFrame(data=similarity_data).sort_values('similarity', ascending=False)
return similarity_dataframe
if __name__ == '__main__':
peptide_backbone = PeptideBuilder(3)
cocktail = Cocktail(peptide_backbone,ligand_library = ['Br', 'F', 'I'])
combinations = cocktail.shake()
print (combinations)
# Render to molecules
molecules = [Chem.MolFromSmiles(x) for x in combinations]
# Render to fingerprints
fingerprints = [FingerprintMols.FingerprintMol(x) for x in molecules]
print(compare_tanimoto_fingerprints_pairwise(smiles=combinations, fingerprints=fingerprints))